Programmatically interacting with the new compounds API
At Strateos we're passionate about frictionless transfers between digital representations and the real physical world. We want to continue to enable our users to blend both their computational workflows with real physical experimentation workflows.


At Strateos we're passionate about frictionless transfers between digital representations and the real physical world. With a highly unique robotic lab that can be programmed we want to continue to enable our users to blend both their computational workflows with real physical experimentation workflows. With the coming launch of multiple chemistry capabilities on the Strateos Robotic Cloud Lab I wanted to show what is possible with the new Compounds API which is currently in preview (As of Jan 2020), showing a few examples of using the API then an example of using it in a workflow along with a pipeline of cheminformatics.
Individual compound records
Let's start by fetching an individual compound record, for this we'll need to know the compound_id. You can get this from the record via the Compounds section of the web application, or by fetching the whole compound list, which we'll do in a moment. Below is a ruby snippet for fetching compound cmp123456.
require 'uri'
require 'net/http'
require 'openssl'
# Set the compound_id of interest
compound_id = "cmp123456"
# Set your organization_id
querystring = "filter[organization_id]=org1235"
url = URI("https://secure.transcriptic.com/api/compounds/?#{id}/#{querystring}")
http = Net::HTTP.new(url.host, url.port)
http.use_ssl = true
http.verify_mode = OpenSSL::SSL::VERIFY_NONE
request = Net::HTTP::Get.new(url)
# Set your API credentials
request["x-user-email"] = 'chell@aperturescience.com'
request["x-user-token"] = 'apk12345678'
response = http.request(request)
puts response.read_bodyLet's inspect the response. You can see that the compound record returned has a number of attributes, including some calculated molecular properties and multiple different identifiers.
{
"data": {
"id": "cmpl1d8yn2kvfwzv4",
"type": "compounds",
"links": {
"self": "https://secure.transcriptic.com/api/compounds/cmpl1d8yn2kvfwzv4"
},
"attributes": {
"name": null,
"reference_id": null,
"organization_id": null,
"created_by": "ad17h37hcyb6uc",
"created_at": "2019-06-17T17:19:13.726-07:00",
"properties": {},
"groups": [],
"search_score": null,
"clogp": "2.3997",
"formula": "C14H13N2+",
"inchi": "InChI=1S/C14H12N2/c1-16-12-8-4-2-6-10(12)14(15)11-7-3-5-9-13(11)16/h2-9,15H,1H3/p+1",
"inchi_key": "SQFZLHHJWJODCA-UHFFFAOYSA-O",
"molecular_weight": "209.27",
"morgan_fingerprint": "iAAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA\nAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEAAAAAAAAAAEAAAAAAAAAAAAAAAAAAAAAAAAAA\nAAAAAAAAAAAAAAAAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQAAAA\nAAAAEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA\nAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA\nAAAAAAEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgAAAAAAAAAAAAAA\nAAAAAAAAAAAAAAAAAAAAAAAAAAAgAAAAAAAAAEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA\nAAAAAAAAAAAAAABAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA\nAAAAAAAAAAAAAAAAAAAAAIAAAAAAAAAAAAAAAAAAAAAABAAAAAAABAAAAAAAAAAAAAAAAAAAAAA=\n",
"sdf": "\n RDKit 2D\n\n 16 18 0 0 0 0 0 0 0 0999 V2000\n 3.0000 -2.5981 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n 1.5000 -2.5981 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0\n 0.7500 -1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n 1.5000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n 0.7500 1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n -0.7500 1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n -1.5000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n -0.7500 -1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n -1.5000 -2.5981 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n -3.0000 -2.5981 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0\n -0.7500 -3.8971 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n -1.5000 -5.1962 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n -0.7500 -6.4952 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n 0.7500 -6.4952 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n 1.5000 -5.1962 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n 0.7500 -3.8971 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0\n 1 2 1 0\n 2 3 2 0\n 3 4 1 0\n 4 5 2 0\n 5 6 1 0\n 6 7 2 0\n 7 8 1 0\n 8 9 2 0\n 9 10 1 0\n 9 11 1 0\n 11 12 2 0\n 12 13 1 0\n 13 14 2 0\n 14 15 1 0\n 15 16 2 0\n 16 2 1 0\n 8 3 1 0\n 16 11 1 0\nM CHG 1 2 1\nM END\n",
"smiles": "C[n+]1c2ccccc2c(N)c2ccccc21",
"tpsa": "29.9"
}
}
}Below is the schema for a Compound record. At its top level its contained in an object called data but most of the details of the compound are located in the attributes object. You can see more record schemas in the API documentation.
[Object] data
If fetching a list of compounds this field is an array of Compound Objects
String data[].id
String data[].type
Object data[].links
String data[].links.self
Object data[].attributes
String data[].attributes.name
String data[].attributes.reference_id
String data[].attributes.organization_id
String data[].attributes.created_by
String data[].attributes.created_at
Object data[].attributes.properties
[String] data[].attributes.groups
This field is an array of Strings for labelling the compounds
String data[].attributes.search_score
This field is used during a similarity search and is between 0 and 1
String data[].attributes.clogp
String data[].attributes.formula
String data[].attributes.inchi
String data[].attributes.inchi_key
String data[].attributes.molecular_weight
String data[].attributes.morgan_fingerprint
String data[].attributes.sdf
String data[].attributes.smiles
String data[].attributes.tpsaFetching your compound list
For fetching your entire compound list, your url should be of the form https://secure.transcriptic.com/api/compounds/?filter[organization_id]=org123 The organization_id filter is required here. And here it is in use in ruby.
require 'uri'
require 'net/http'
require 'openssl'
querystring = "filter[organization_id]=org123"
url = URI("https://secure.transcriptic.com/api/compounds/?#{querystring}")
http = Net::HTTP.new(url.host, url.port)
http.use_ssl = true
http.verify_mode = OpenSSL::SSL::VERIFY_NONE
request = Net::HTTP::Get.new(url)
request["x-user-email"] = 'chell@aperturescience.com'
request["x-user-token"] = 'apk12345678'
response = http.request(request)
puts response.read_bodyThis will then return an array containing compound objects:
{"data":[{"id": "1", ...},
{"id": "2", ...},
{"id": "3", ...},
...
]
}Creating a new compound
Let's now move on to creating compounds in Strateos, let's take this molecule below:

CC12CCC(O)CC1CCC1C2CCC2(C)C(c3ccc(=O)oc3)CCC12OHere we create a POST request to create this new compound. Compounds can be created from sdf, SMILES and InChi representations. You need to only provide one identifier in the body of the request. In the example below we're using the SMILES representation.
require 'uri'
require 'net/http'
require 'openssl'
url = URI("https://secure.transcriptic.com/api/compounds")
http = Net::HTTP.new(url.host, url.port)
http.use_ssl = true
http.verify_mode = OpenSSL::SSL::VERIFY_NONE
request = Net::HTTP::Post.new(url)
request["accept"] = 'application/json'
request["content-type"] = 'application/json'
request["x-user-email"] = 'chell@aperturescience.com'
request["x-user-token"] = 'apk12345678'
request.body = "{\"data\":
{\"attributes\":
{\"compound\":
{\"smiles\":\"CC12CCC(O)CC1CCC1C2CCC2(C)C(c3ccc(=O)oc3)CCC12O\"}
}
}
}"
response = http.request(request)
#> Returns a 201 success
puts response.read_bodyLet's now inspect the 201 Success response from Strateos:
Using Python and the RDKit with the Strateos API
We're going to switch over from Ruby to Python now so we can make use of the RDKit. In this example we're writing a small function that can fetch a compound from your Strateos collection by its Strateos compound_id then we use the RDKit to create an RDKit Mol object. From this point we can do any downstream manipulations or analyses that we would use the RDKit for, in this example just returning the number of atoms in the compound.
from rdkit import Chem
import requests
import json
def MolFromStrateos(compound_id):
url = "https://secure.transcriptic.com/api/compounds/" + compound_id
print(url)
# Set your organiation ID in the filter
querystring = "filter[organization_id]=org1235"
# Set your API credentials in the headers
headers = {
'x-user-email': "chell@aperturescience.com",
'x-user-token': "apk1234667"
}
response = requests.request("GET", url, headers=headers, params=querystring}
try:
mol = Chem.MolFromInchi(response.json()["data"]["attributes"]["inchi"])
except ValueError:
try:
mol = Chem.MolFromSmiles(response.data.smiles)
except ValueError:
print("Error parsing compound from Strateos")
pass
return mol
test_mol = MolFromStrateos("cmpl1d352345fwzv4")
print(test_mol.GetNumAtoms())
# => 8Let's combine this function along with a cool code example from the RDKit blog written by Greg Landrum that was designed to show off some of the new drawing features. Below we will fetch a molecule from Strateos, create and RDKit Mol object, generate some conformers and look at partial charge variation across those conformers.
from rdkit.Chem import rdEHTTools
from rdkit.Chem import rdDistGeom
my_mol = MolFromStrateos("cmpl1dms9rg9azcrv")
mh = Chem.AddHs(my_mol)
ps = rdDistGeom.ETKDGv2()
ps.pruneRmsThresh = 0.5
ps.randomSeed = 0xf00d
rdDistGeom.EmbedMultipleConfs(mh,10,ps)
print(f'Found {mh.GetNumConformers()} conformers')
chgs = []
for conf in mh.GetConformers():
_,res = rdEHTTools.RunMol(mh,confId=conf.GetId())
chgs.append(res.GetAtomicCharges()[:my_mol.GetNumAtoms()])
chgs = np.array(chgs)
mean_chgs = np.mean(chgs,axis=0)
std_chgs = np.std(chgs,axis=0)
d = Draw.MolDraw2DSVG(800, 800)
SimilarityMaps.GetSimilarityMapFromWeights(my_mol,list(mean_chgs),draw2d=d)
d.FinishDrawing()
SVG(d.GetDrawingText())After generating the partial charge distributions for 10 conformers, we generate a map of the mean partial charges across the molecule of interest. RDKit can generate these awesome visualizations.
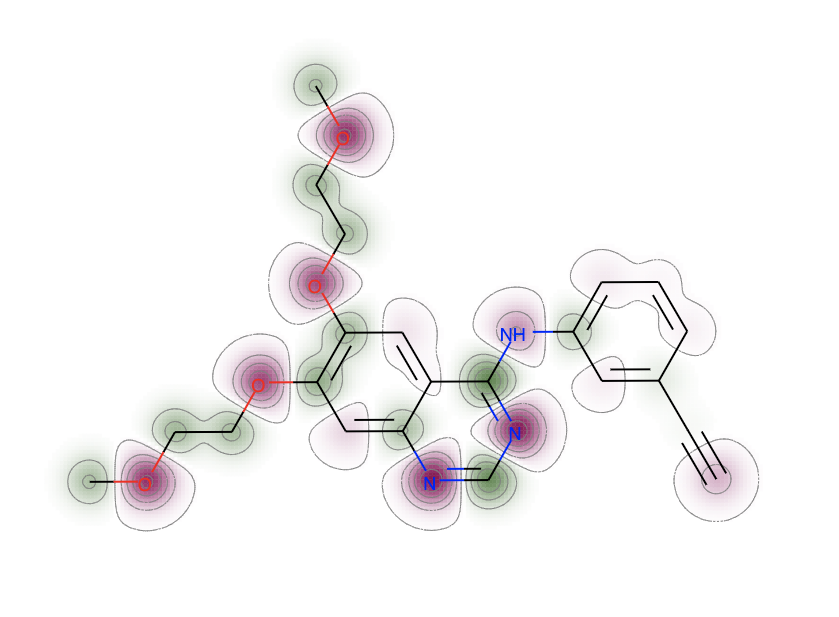
Next rather than looking at the mean partial charge distribution for the conformers, we want to look at the standard deviation of partial charges across all conformers.
print(std_chgs)
print(max(std_chgs),min(std_chgs))
d = Draw.MolDraw2DSVG(800, 800)
SimilarityMaps.GetSimilarityMapFromWeights(my_mol,list(std_chgs),draw2d=d)
d.FinishDrawing()
SVG(d.GetDrawingText())
#=> [0.00685736 0.00529414 0.00595085 0.00377577 0.00757177 0.01016299
#0.00893538 0.00665945 0.00847515 0.0084792 0.00717066 0.01004357
#0.01300254 0.01362973 0.00665798 0.00511058 0.00900267 0.00652139
#0.00640103 0.01126982 0.00882498 0.01722642 0.02212623 0.01361278
#0.07565581 0.02092477 0.01805508 0.03461893 0.05119605]
#0.07565581351596404 0.0037757653659223132
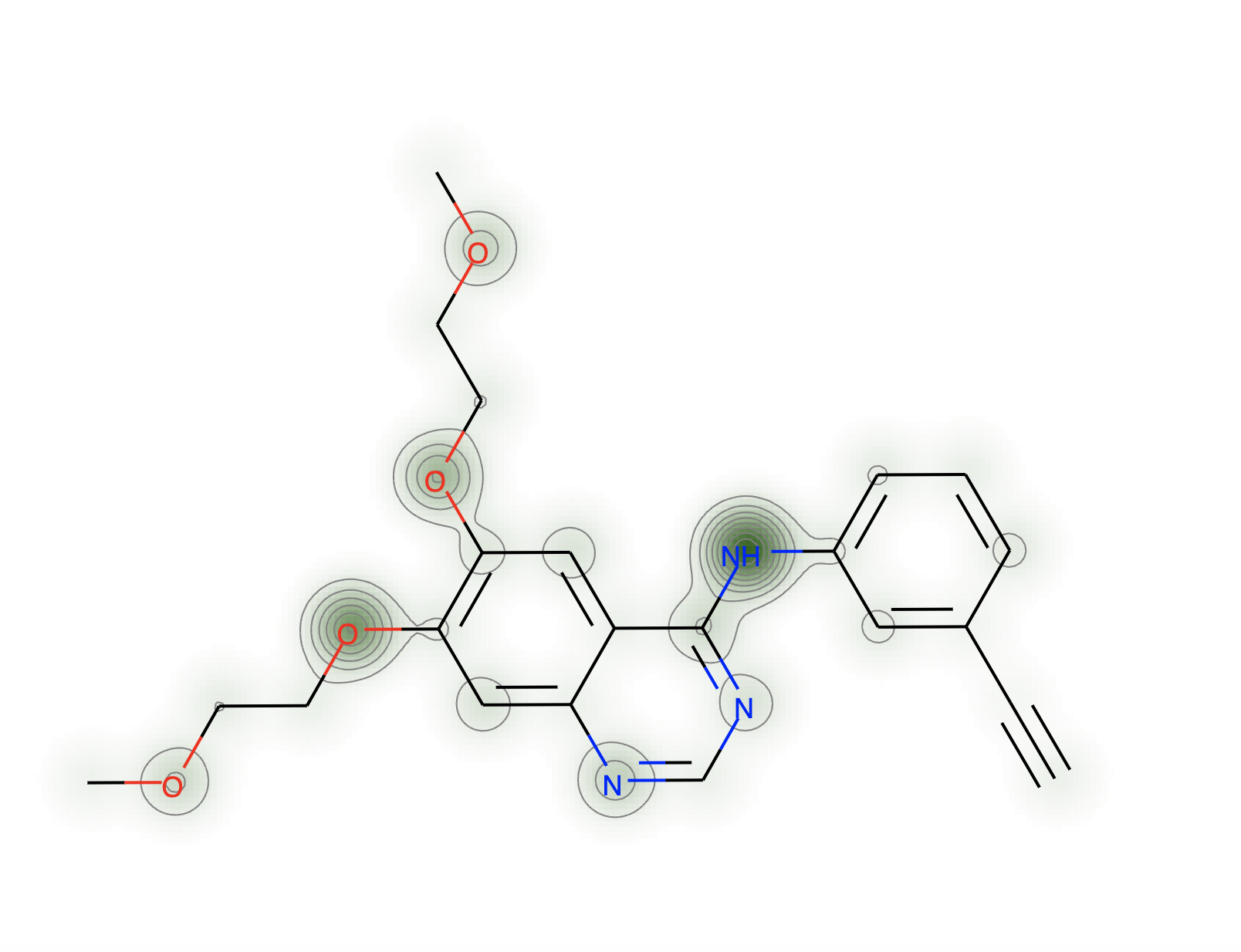
Creating Strateos compounds from the RDKit
Let's take one final step to create a function to make it easier to go from the RDKit Mol objects to creating them on Strateos. In this function we wrap a Requests POST object and construct the payload of the request from function arguments including a Mol object as the primary argument. We use Chem.MolToSmiles() to get the SMILES representation of the molecule and send this in the payload. We also populate the labels field to tag the molecule as a statin, so it will be grouped with all other compounds tagged statin on Strateos.
import json
import requests
organization_id = "org123"
def MolToStrateos(mol, labels=[]):
url = "https://secure.transcriptic.com/api/compounds/"
headers = {
'x-user-email': "chell@aperturescience.com",
'x-user-token': "user-token"
}
data = {"data":
{"attributes":
{"compound":
{"smiles": Chem.MolToSmiles(mol)},
"organization_id": organization_id,
"groups": labels
}
}
}
response = requests.request("POST", url, json=data, headers=headers)
return response
response = MolToStrateos(rosuvastatin, ["statin"])
#> <Response [200]>Then if we login to our Strateos account we can see our creation of rosuvastatin.
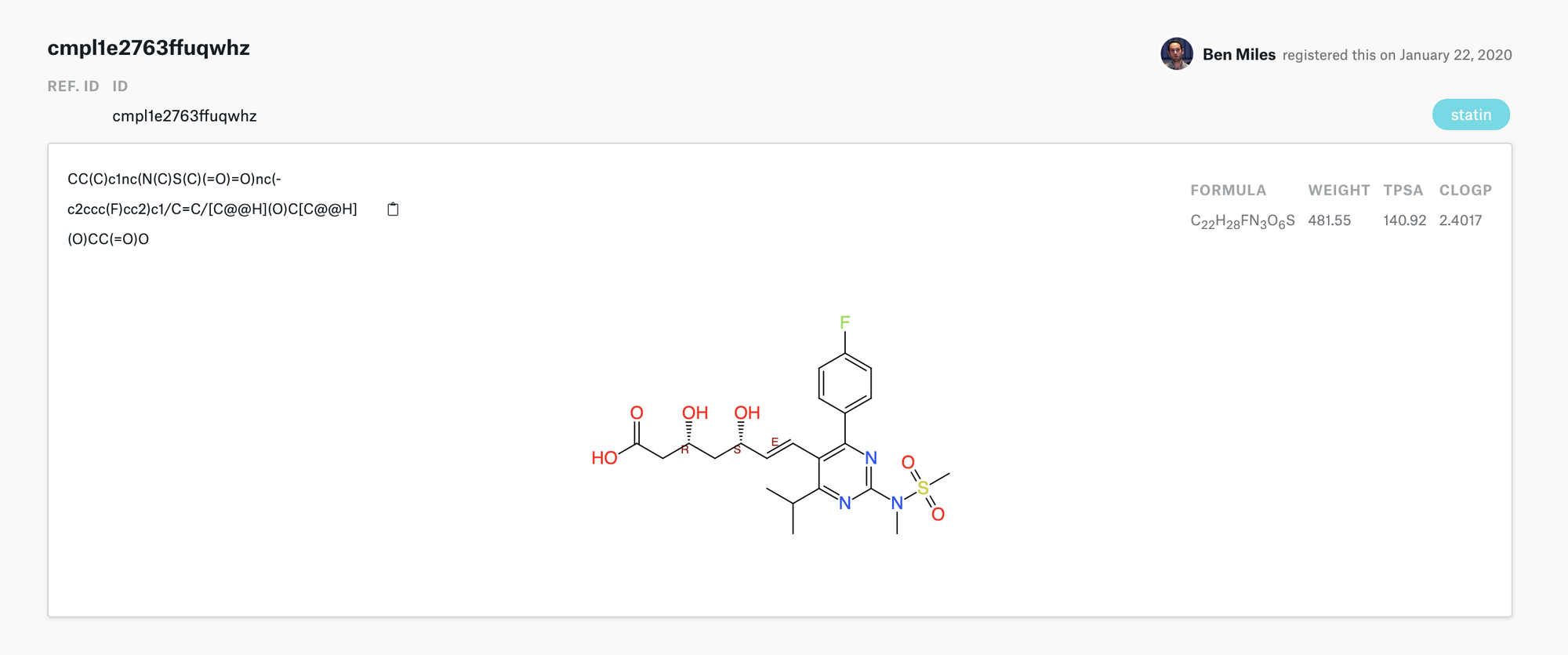
One could use this in a workflow of enumeration where you want to generate 10's of variations of a molecule then create them on Strateos all tagged the same way so they can be identified as a group.
Recap
In this post we went through fetching both individual and lists of compound records from the Strateos API using both Ruby and Python. We also walked through the structure of the Compound object. Finally we went through a couple of examples combining the awesome cheminformatics package the RDKit with the Strateos compounds endpoint, to integrate Strateos into a cheminformatics pipeline. This article should given an indication of how one will be able to move seamlessly from cheminformatic pipelines through the synthesis and characterization of molecules using the Strateos Robotic Cloud Lab.